


Key areas
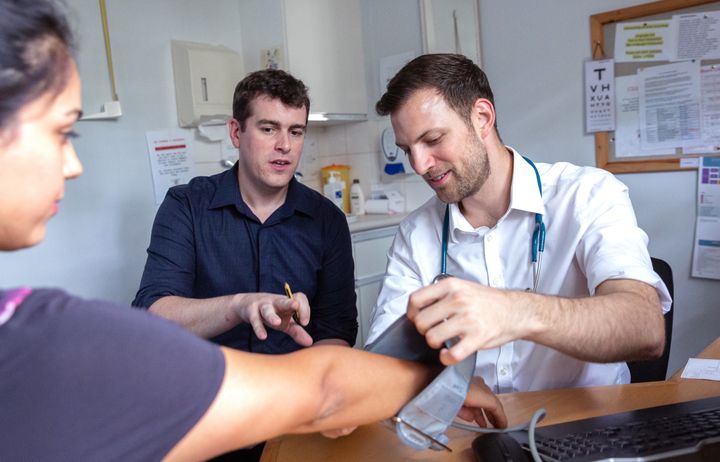
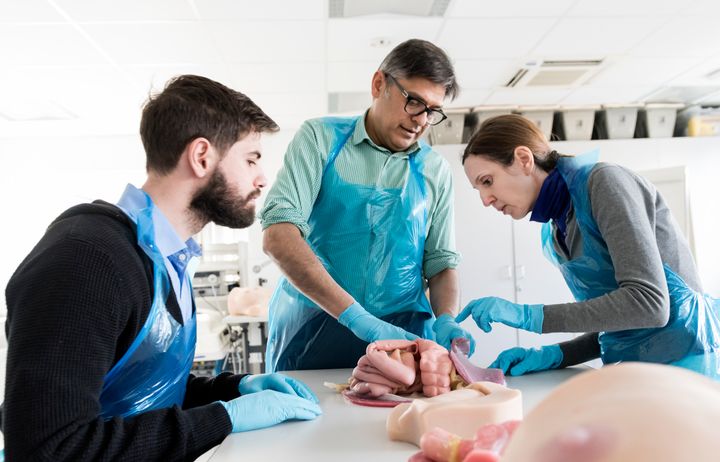

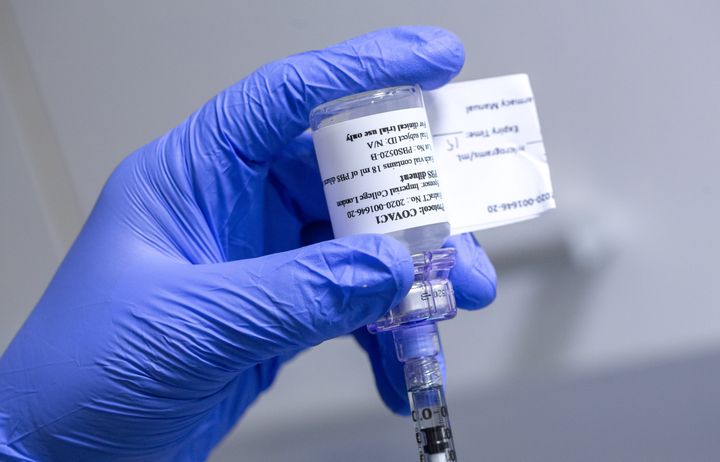

Spotlight on postgraduate programmes
Our Master’s programmes draw on recognised excellence in research to offer a wide range of programmes for careers in lab-based science, public health and clinical healthcare.

Imperial academic wins €2.4m European funding to improve solar harvesting tech

Meningococcal vaccine is cost effective at protecting men against gonorrhoea

Shape-shifting cancer cell discovery reveals potential skin cancer drug targets
More NewsOur Master’s programmes draw on recognised excellence in research to offer a wide range of programmes for careers in lab-based science, public health and clinical healthcare.



